Databases and Alignment
Name: Aadam
Class: INFO-B 519
Due Date: September 19th, 11:59pm
Purpose: The purpose of this assignment is twofold: first, to understand and utilize common biological data databases, and second, to get experience with performing sequence alignment, both by hand and with common alignment tools.
Skills
In order to complete this assignment, you will need to
- utilize common biological databases
- understand the redundancy rules for common biological databases
- use critical thinking to determine the optimal sequence alignment
- utilize common alignment tools
Knowledge
This assignment will help you gain the following:
- how to navigate and use common biological databases
- how to perform sequence alignment using dynamic programming
- how to navigate and use common alignment tools %%
Based on the accession number/ID of each biological sequence list below, give its sequence database name, type (DNA/RNA/protein), and sequence length? (16 points)
ID Database Sequence Type Length M20778 GenBank DNA 1008 NP_000537 NCBI Protein 393 UHJ46542 GenBank Protein 322 NM_028151 NCBI RNA 3309 5YM7 PDB Protein 1.56 P01911 UniProt Protein 266 DF915701 GenBank DNA 1976
What are the rules for removing redundant sequences for the following databases: RefSeq, Swiss-Prot, TrEMBL, UniRef100, UniParc? (24 points).
RefSeq:1 - When two RefSeq sequences are found to be redundant, one is retained as primary, and the other becomes secondary. - If the redundant sequences were associated with different Gene records, those records are merged into a single record.
TrEMBL: 2 - UniProtKB/TrEMBL maintains non-redundancy for identical, full-length protein sequences within the same species. - Fragments, isoforms, variants, etc., encoded by the same gene are stored in separate entries.
Swiss-Prot: 3 - UniProtKB/Swiss-Prot ensures non-redundancy for all protein products encoded by one gene in a given species within a single record. This includes isoforms, fragments, genetic variations, and more. - When different genes in the same species produce the same protein sequence, they are merged into a single UniProtKB/Swiss-Prot record, listing the gene names in the gene name subsection. - Identical sequences from different species are stored in different records. - Manual annotation involves merging UniProtKB/TrEMBL entries corresponding to protein products encoded by the same gene and documenting differences.
UniParc: 4 5 - UniParc is a non-redundant database storing identical protein sequences in a single record, regardless of the species. - Each record has a unique identifier (UPI), ensuring uniqueness across different source databases. - It tracks sequence changes in source databases and maintains a history of these changes.
UniRef100: 6 7 - UniRef100 is generated by clustering identical sequences and subfragments with 11 or more residues from any organism into a single UniRef entry. - It combines identical sequences and subfragments, providing a representative protein sequence along with accession numbers of all merged entries.
These rules aim to minimize redundancy while ensuring the representation of distinct sequences and variations in biological sequence databases. The specifics may evolve over time with updates and manual curation efforts, especially for Swiss-Prot and TrEMBL, to provide accurate and comprehensive information to researchers.
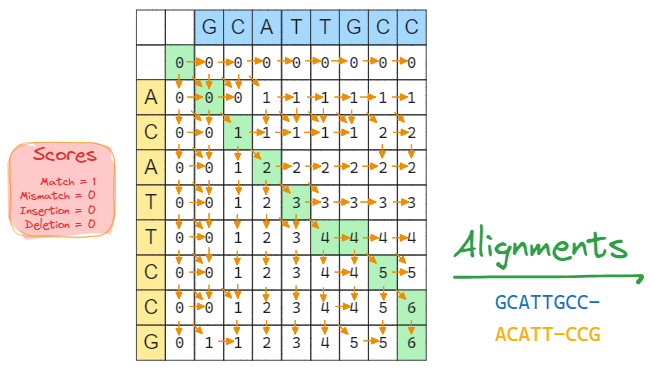
With the following similarity scoring scheme: score(match) = 1, score(mismatch) = 0, score(deletion) = 0, score(insertion) = 0, use the dynamic programming method to manually compute a best alignment of two sequences S1 = GCATTGCC and S2 = ACATTCCG. (The dynamic programming table, backtracking process and final alignment are needed). (30 points)
Find the protein sequences: P63279 and P61916 from biological sequence databases, and use “EMBOSS needle” to compute the optimal global alignment of the two sequences. What are the score and identity of the optimal GLOBAL alignment for each of the following similarity matrices: PAM30, PAM70, BLOSUM62, BLOSUM80 (using default gap opening penalty 10 and gap extension penalty: 0.5). (20 points)
Matrix Score Identity PAM30 7.5 25/267 (9.4%) PAM70 18.5 28/250 (11.2%) BLOSUM62 29.0 24/240 (10.0%) BLOSUM80 89.0 17/244 (11.1%)
Use “EMBOSS water” to compute the optimal local alignment of the two sequences P63279 and P61916. What are the score and identity of the optimal LOCAL alignment for each of the following similarity matrices: PAM30, PAM70, BLOSUM62, BLOSUM80 (using default gap opening penalty 10 and gap extension penalty: 0.5). (10 points)
Matrix Score Identity PAM30 34.5 8/29 (27.6%) PAM70 31.0 17/50 (34.0%) BLOSUM62 40.0 16/46 (34.8%) BLOSUM80 96.5 33/175 (18.9%)